[ad_1]
Some molecules tend to bond closely to the ice surface, forming a curved interface that inhibits further growth of the ice crystal.

Some plants, insects and aquatic creatures include protein molecules of this type that act as natural antifreeze agents, which allow organisms to withstand freezing temperatures.
In Journal of chemical physics, from AIP Publishing, a team of researchers has described a computational technique for modeling ice bonding, which involves using a polarization method to induce ice formation in the simulation.
Antifreeze proteins work by binding to an existing interface between liquid water and ice. The resulting curved surface prevents ice growth. Additionally, there are ice-nucleating molecules that catalyze the development of ice from super-cooled liquid water.
Both phenomena require insights into how molecules bind to ice. Gaining knowledge about ice bonding is essential for applications ranging from climate modeling to organ cryopreservation. However, at present, there are no computational techniques to efficiently model this phenomenon.
The central advantage of the ice biasing simulation approach is that it simultaneously identifies the bonding surface of the ice, the face of the ice it binds to, and the bonding mode.
Valeria Molinero, Author of the study, University of Utah
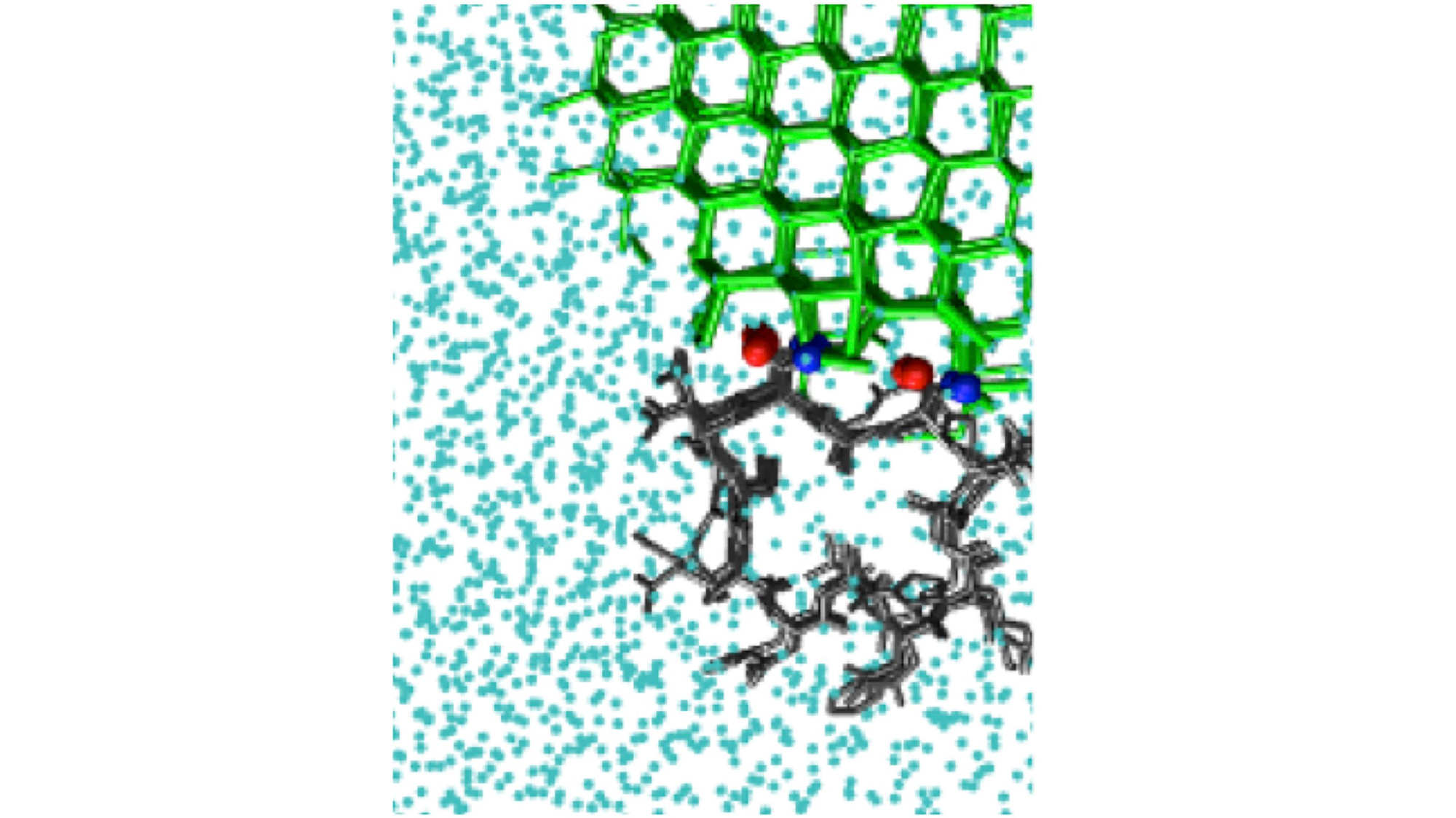
Two types of models were made by the investigators. One is a fully atomic model that includes all atoms present in the ice and liquid phases of water, as well as in the antifreeze type molecule.
The second model studied is known as a coarse-grained model, which reduces the computational resources used by mixing atoms into simpler structures.
The study analyzed several ice-binding molecules, such as polyvinyl alcohol, a synthetic inhibitor of ice recrystallization, and natural antifreeze proteins, such as beetle. Tenebrio molitor.
Since proteins have very small surfaces that bind to ice, they are difficult to simulate. This limits the size of the ice crystal that can be bound by them.
Some systems have more than one location where ice binds. This applies to the natural antifreeze protein found in the sea ice diatom The Frailariopsis cylinder. The researchers designed a technique called “cap and repeat” to identify if a protein like this has more than one ice-binding surface (IBS).
In this strategy, we first ran a biased simulation to detect an IBS. Then, we cover the IBS to prevent ice from forming on it and run a second polarization simulation to find out if ice forms at other sites..
Valeria Molinero, Author of the study, University of Utah
The techniques developed as part of the study show enormous potential for several applications, such as determining molecules to safeguard frozen tissue at the time of storage, gaining further insights into natural antifreeze proteins, as well as in climate models, where ice in the air plays a vital role.
Journal reference:
Naullage, PM, et al. (2020) Computationally Efficient Approach to Identifying Ice-Binding Surfaces and How Ice Binds. The Journal of chemical physics. doi.org/10.1063/5.0021631.
Source: https://www.aip.org/
Source link
